25 Diciembre 2020
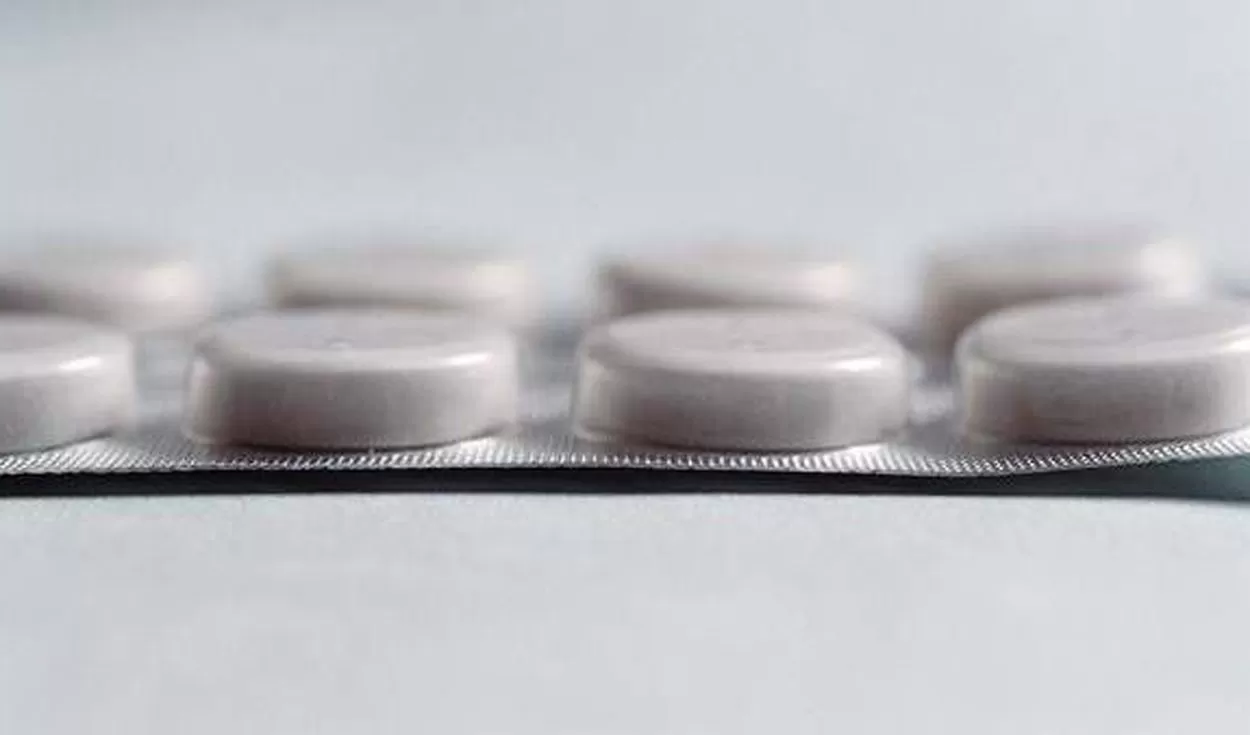
La Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (Anmat) dispuso ayer mediante una resolución la "suspensión preventiva" y "la comercialización en todo el territorio nacional" de la ranitidina para uso por vía oral, siguiendo la línea de organismos internacionales especializados en la investigación de medicamentos.
Se trata de un producto prescripto para casos de gastritis, de reflujo gástrico y otras afecciones estomacales similares. Por ello, suele ser recetado para los pacientes que se exceden en las fiestas con alimentos o bebidas. Pero en esta Navidad y Año Nuevo, según la Anmat, esa droga no debe ser comercializada.
La suspensión preventiva alcanza "todas las especialidades medicinales que contengan ranitidina como Ingrediente Farmacéutico Activo (IFA), como monodroga o en asociación con otros IFA, en sus formas farmacéuticas orales, en todas sus concentraciones y presentaciones, por los fundamentos expuestos en el considerando de la presente disposición".
"En sus Novedades Internacionales y Nacionales en Seguridad de Medicamentos del mes de septiembre de 2020, el Departamento de Farmacovigilancia y Gestión de Riesgo informó que el Comité de Medicamentos de Uso Humano (CHMP) Agencia Europea de Medicamentos (EMA) había confirmado su recomendación de suspender todos los medicamentos que contienen ranitidina en la Unión Europea debido a la presencia de bajas concentraciones de la impureza denominada N-nitrosodimetilamina (NDMA), y que se había hallado NDMA en varios medicamentos que contienen ranitidina por encima de las concentraciones consideradas aceptables, existiendo preguntas sin resolver acerca de la fuente de esta impureza y alguna evidencia de que la NDMA podría formarse a partir de la degradación de la ranitidina misma, con concentraciones crecientes durante su vida útil, sin estar claro si la NDMA podría también formarse dentro del organismo", señaló la Anmat en la Disposición 9209/2020, publicada ayer.
Ante esta situación, el Departamento de Farmacovigilancia y Gestión de Riesgo consideró "necesario suspender preventivamente la comercialización de las formas farmacéuticas de administración oral de las especialidades medicinales con ranitidina y proceder a su retiro del mercado" en un plazo de 30 días.
Textuales de la disposición de la ANMAT
1) "En octubre de 2019 un comunicado de esta ANMAT informó a la población que, luego de haber analizado la evidencia científica disponible bajo los principios de precaución e incertidumbre del ingrediente farmacéutico activo ranitidina, no se había hallado entonces evidencia suficiente para sugerir la suspensión de los tratamientos con la misma y que la Administración se encontraba trabajando y monitoreando el tema, con el fin de establecer los lineamientos necesarios para fortalecer los procesos de control y evitar la presencia de la impureza NDMA en las especialidades medicinales que contengan el ingrediente farmacéutico activo mencionado y/o establecer los niveles aceptables de seguridad".
2) "En el comunicado e informó que hasta tanto se contara con información fehaciente acerca del contenido de la impureza NDMA en medicamentos que contengan ranitidina como principio activo, se suspendería preventivamente la elaboración de nuevos lotes en formas farmacéuticas orales por parte de los titulares de producto, que se sugería no interrumpir los tratamientos y que, en caso de existir alguna duda, era el profesional médico quien debía evaluar la alternativa terapéutica existente en el mercado apropiada para cada paciente en particular".
2) "La EMA, en abril de 2020, refirió que existía alguna evidencia de que la NDMA podía formarse a partir de la degradación de la ranitidina misma, con concentraciones crecientes durante su vida útil, que no estaba claro si la NDMA podía también formarse dentro del organismo y que el Comité de Medicamentos de Uso Humano (CHMP) había recomendado suspender preventivamente todos los medicamentos que contenían ranitidina en la Unión Europea".
3) "También en abril de 2020, la Administración de Medicamentos y Alimentos (FDA) de los Estados Unidos de América comentó que nuevas evaluaciones impulsadas en otros laboratorios confirmaron que las concentraciones de NDMA se incrementaban en la ranitidina incluso bajo condiciones normales de almacenamiento y se había hallado que la NDMA se incrementaba significativamente en muestras almacenadas a mayores temperaturas, incluyendo temperaturas a las que el producto podía estar expuesto durante la distribución y la manipulación por los consumidores".
4) "Según dicha agencia los tests también mostraron que cuanto más antiguo era el producto con ranitidina, o mayor el tiempo desde que se había fabricado, mayor era la concentración de NDMA y que estas condiciones podían elevar la concentración de NDMA por encima del límite de ingesta diaria admisible".
Temas
Anmat
Lo más popular



















